


Advances: Protein folding with artificial intelligence from DeepMind's AlphaFold2
Computing nature’s most complicated biomolecules

The “Advances” series of articles discuss technological breakthroughs in the life sciences. These articles present important findings from scientific studies and other resources.
Highlights and takeaways
Atomic resolution of intramolecular and intermolecular interactions are highly sought after to better understand biological processes and aid drug development.
DeepMind’s AlphaFold2 outperformed all other groups at the protein structure prediction competition known as CASP. The assessment results are briefly evaluated.
Contrary to mainstream media, protein folding is not yet a solved problem. However, deep learning algorithms have accelerated our ability to accurately predict structures for well-behaved proteins.
There has been so much excitement regarding protein structure prediction and design. I am personally very fascinated by recent developments and have an optimistic outlook for the future of protein science. By popular request, this article discusses protein folding and the recent CASP14 results of AlphaFold2 from DeepMind.
The relationship between sequence, structure, and function
Proteins are macromolecules responsible for numerous biological processes. They are linear chains of amino acids folded in a manner which enables a particular function, such as mediating molecular interactions or catalyzing chemical reactions. Characterizing protein structures can help with discerning how these macromolecules facilitate biological activity. The composition of a protein can be understood in four main categories: primary structure, secondary structure, tertiary structure, and post-translational modifications. The primary structure is the precise sequence of amino acids. In living organisms, the primary structure is encoded by nucleic acids and synthesized by translational machinery. The secondary structure is the three dimensional arrangement of a local segment, which are often α-helices or β-strands. These are driven by hydrogen bonding across the backbone of the polypeptide. The tertiary structure is the collective three dimensional configuration of the polypeptide chain and its secondary structures. Sometimes, proteins will assemble into larger complexes and this is known as the quaternary structure. Post-translational modifications are changes to the protein following normal biosynthesis, and often mediated by enzymes.
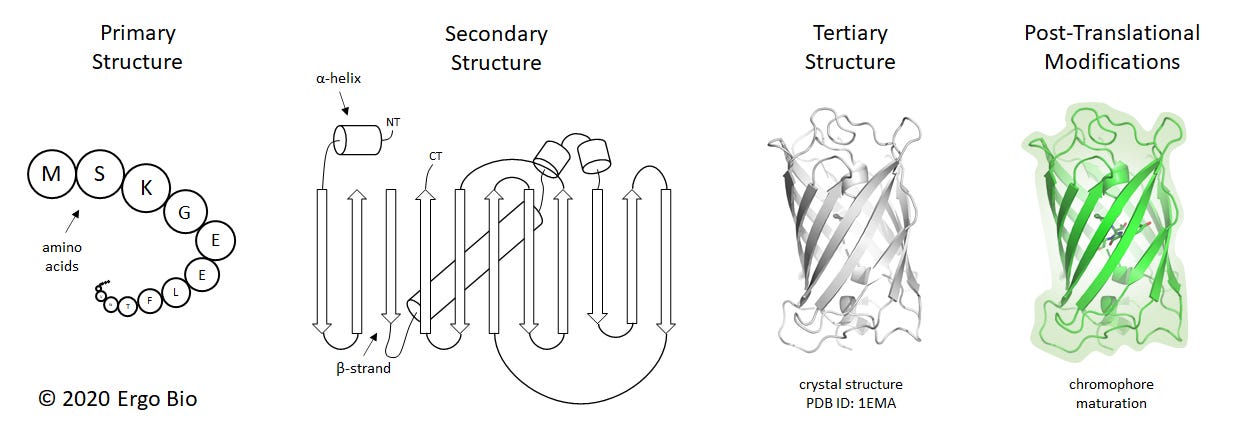
These structural categories can be described for a protein such as the green fluorescent protein (GFP), which was first determined at 1.9 Å resolution in 1996 (ref). GFP comprises of 238 residues (primary structure) and forms 4 α-helices and 11 β-strands (secondary structure). GFP is a β-barrel (tertiary structure) and spontaneously forms a chromophore from residues S65-Y66-G67 (post-translational modification). GFP is the protein responsible for fluorescence in the jellyfish Aequorea victoria and has numerous applications as a genetically-encoded reporter.

Source: Dobson 2003
Protein folding has been the topic of rigorous research for decades. Christian B. Anfinsen postulated protein structures are primarily governed by amino acid sequences (ref). Cyrus Levinthal hypothesized that proteins spontaneously and quickly fold along a pathway guided by local interactions (ref). Energy landscapes are a conceptual and quantitative way to understand how proteins fold and form structures (ref). The native configuration of a folded protein is typically the structure which is most thermodynamically stable in physiological conditions. During and after synthesis, polypeptides will fold and transition across states, eventually arriving at the native configuration. However, it is important to recognize that the native configuration is actually an ensemble of states described by a distribution across the energy landscape. Furthermore, the native configuration is that which is accessible through a series of transitions (i.e. the folding pathway). If there exists a lower energy state, the protein may not reach it if there are no intermediate states that can be reasonably sampled. To help traverse the energy landscape, living organisms have developed molecular chaperones and subcellular compartments to guide nascent polypeptides towards their final destination. Sometimes, chemically synthesizing proteins will not have the same configuration as those that are made through biosynthesis.
Computing protein structure from amino acid sequence

Source: Senior et al. 2020
Critical Assessment of Structure Prediction (CASP) is a biennial, global contest for research groups to test their methods for structure prediction (ref). DeepMind first entered the competition in its 13th round and surprised the world with its deep learning system known as AlphaFold (ref). Briefly and simply, AlphaFold is a convolutional neural network (CNN) trained on existing protein structures to predict pairwise distances between residues (ref). Homologous sequences are assembled as a multiple sequence alignment (MSA) with a sliding window rather than by segmentation. Covariation features are extracted from the MSA in a manner like that of image recognition. A discrete probability distribution is calculated for each residue within a 64x64 convolutional block of the distance matrix. Collectively, this describes the distance histogram (distogram) and forms a potential by which gradient descent can be deployed to optimize backbone torsion angles. The structure is then relaxed using Rosetta, a Monte Carlo minimization procedure for protein structure prediction and design (ref).
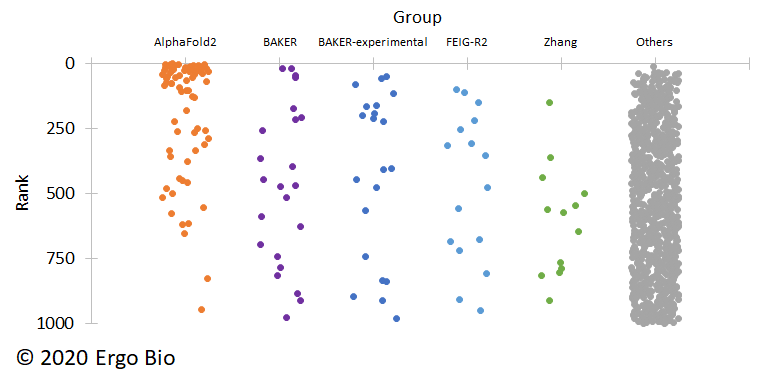
This year, in the 14th round, DeepMind yet again exceeded expectations with AlphaFold2. DeepMind altered the algorithm to better address global structural constraints instead of focusing on pairwise coevolution, although details have yet to be released (ref). It takes a bit of diligence, but the submissions are publicly available and can be freely analyzed (CASP14). Compared with other top research groups, AlphaFold2 had exceedingly more submissions that scored in the top 1000 and stacked the upper rankings.

To better understand the results, here are a couple notes: CASP assessment is based on an alternative measurement to root-mean-square deviation (RMSD) known as global distance test (GDT) (ref) and CASP targets are classified for template-based modeling (TBM) and free modeling (FM) (ref). Structural “templates” can be identified based on sequence homology, which aids the process for structure prediction. Targets without a detectable template are deemed more challenging to predict. In CASP13, DeepMind’s AlphaFold placed first in the FM category and the Zhang group from the University of Michigan placed first in the TBM category. This year, DeepMind’s AlphaFold2 stands head and shoulders above the rest in both categories.

The best and worst predictions by AlphaFold2, along with real structures, are presented above. I generated chainbow cartoon representations using PyMOL2 from Schrödinger. Oxalyl-CoA decarboxylase from O. formigenes had multiple structure templates that enabled near-perfect prediction to the solve structure, with AlphaFold2 scoring an incredible GDT = 99.07 and RMSD = 0.613 (ranked first). In comparison, Ebsa protein from cyanobacteria did not have any structure template and was difficult to solve, with AlphaFold2 scoring a dismal GDT = 44.60 and RMSD 7.124 (ranked third). Clearly, other research groups also had a challenging time with the structure prediction.

Taking a closer look (with side chains shown), there are a couple interesting configurations in AlphaFold2’s prediction of T1029. Aromatics like Y37 and W60 are packed with very particular coordination of hydrogen bonds with the backbone. A kink in an α-helix appears due to disruptive proline P31. These could be unfavorable in a natural context, and perhaps the future AlphaFold2 publication will discuss why prediction of T1029 was so difficult.
Protein folding is not a solved problem, but we’re doing better
The results of CASP14, and especially that of AlphaFold2, are amazing. Scientists are pushing the boundaries of what is possible and deep learning has helped accelerate our ability to predict protein structures from amino acid sequences. However, headlines from mainstream media falsely indicate that protein folding is a solved problem (ref). Rather, well-behaved proteins, especially those with homologous sequences of known structures, can be accurately predicted. The limited availability of solved structures for certain classes of proteins make prediction challenging. Large and complicated proteins can be difficult to predict, even if smaller fragments have been previously characterized. Even small peptides, such as miniproteins with and without disulfides or irregular protein loops, can be quite challenging to model (ref). Membrane proteins are tough to structurally determine given the lipid/water interface (ref). Fold-switching proteins are often resolved in only one conformation (ref). Thus, there are numerous ways in which protein folding is not yet solved. Despite these outstanding queries, structure prediction has been significantly improved with deep learning algorithms. With time, more complex proteins can be predicted, leading to greater understanding of biology and aiding the development of drugs. Furthermore, this work represents what is possible by R&D efforts in the private sector. DeepMind has done a marvelous job building the AlphaFold technology. Although not discussed, Schrödinger has some wonderful software for molecular modeling as well, including PyMOL2 that I use nearly every day.
Author information
Ergo Bio closely follows innovation in the biotechnology space and evaluates interesting drugs and deals. It is run by Vandon T Duong (LinkedIn), feel free to connect! I am a biotech enthusiast and a molecular engineer by training. I am also an avid consumer of news and research around precision medicine.
Ergo Bio pages
Disclaimer
This article serves informational purposes only and should be treated strictly as educational material, not as investment recommendation or legal advice. The information presented may be inaccurate or out-of-date. The contributing authors and editors disclaim liability for any errors or omissions. Any opinions expressed may change without notice. Ergo Bio LLC reserves all rights to the content generated through this resource hub (Ergo Bio Insights).